Template: Solution-State NMR#
![]()
Solution-state NMR is not much different from solid-state NMR, except that we need to include the correlation time of isotropic tumbling (anisotropic tumbling not fully implemented- contact us if interested in this feature). Compare the files HETs_15N.txt to ubi_soln.txt, where ubiquitin data is from solution-state.
Note that the placeholder data in this template comes from the following paper:
C. Charlier, S.N. Khan, T. Arquardsen, P. Pelupessy, V. Reiss, D. Sakellariou, G. Bodenhausen, F. Engelke, F. Ferrage. Nanosecond time scale motions in proteins revealed by high-resolution NMR relaxometry. J. Am. Chem. Soc. 2013 135, 18665-72
If you intend to run on a local pyDR installation, then you just need to point pyDR to the file. If you want to run in Google Colab, the file needs to be available online somehow. We suggest a shareable weblink (for example, also available in Dropbox, Google Drive, etc.).
Parameters#
Below, you find the parameters you would typically change for your own analysis
#Where's your data??
# path_to_nmr_data='data/ubi_soln.txt' #Data stored locally
path_to_nmr_data='https://github.com/alsinmr/pyDR_tutorial/raw/main/data/ubi_soln.txt' #Github raw link
# path_to_nmr_data='https://drive.google.com/file/d/1U4mGNGyIEH9XNqDvI4qUWQZagPkwi7dx/view?usp=share_link' #Google drive share link
# How many detectors
n=4
# Do you have S2 data in your text file?
inclS2=True
#Is there a PDB ID or saved topology file associated with your structure?
#(set =None if no structure)
topo='1D3Z'
#What Nucleus did you measure? (see below for more explanation)
Nuc='N' #This refers to the backbone nitrogen, specifically
segids=None # Usually, segment does not need to be specified
# SETUP pyDR
import os
os.chdir('../..')
#Imports
import pyDR
Load NMR Data#
The best way to get load your data depends if you’re running locally (just point to the file) or if you’re running online (download from somewhere, mount Google Drive)
Note that you can use commands such as ‘ls’, ‘cd’, and ‘pwd’ if you’re confused where your files are.
data=pyDR.IO.readNMR(path_to_nmr_data)
ls
Bike_stuff/ Grants_etc./ WebEx/
Bike_theft/ Habilitation/ Zoom/
Courses/ Jobs/ jupyter_books/
Covid_vaccine/ JupyterTests/ misc/
DesktopPhotos/ Leipzig_misc_docs/ misc_UniLeipzig/
Dynamics/ Login_infos/ python/
DynamicsLaptop@ MATLAB/ website/
EM2020/ Manuals/ website201113/
ETH/ Microsoft User Data/ website211025/
EndNote/ PassportCRBA/ website230221/
EndNoteLibraries/ ROSETTA/
GitHub/ Travel/
Put data into a project#
Projects are convenient ways to manage a lot of data, and provide convenient tools for overlaying data in 2D plots, as well as visualizing data in 3D in ChimeraX. Not used extensively in this template.
proj=pyDR.Project(directory=None) #Include a directory to save the project
proj.append_data(data)
Attach structure to the data#
Download a pdb and attach it to the data object. For protein backbone dynamics, we recommend using the labels to provide the residue number, making this step easier.
Available bonds (‘Nuc’)
N,15N,N15 : Backbone N and the directly bonded hydrogen
C,CO,13CO,CO13 : Backbone carbonyl carbon and the carbonyl oxygen
CA,13CA,CA13 : Backbone CA and the directly bonded hydrogen (only HA1 for glycine)
CACB : Backbone CA and CB (not usually relaxation relevant)
IVL/IVLA/CH3 : Methyl groups in Isoleucine/Valine/Leucine, or ILV+Alanine, or simply all methyl groups. Each methyl group returns 3 pairs, corresponding to each hydrogen
IVL1/IVLA1/CH31 : Same as above, except only one pair
IVLl/IVLAl/CH3l : Same as above, but with only the ‘left’ leucine and valine methyl group
IVLr/IVLAr/CH3r : Same as above, but selects the ‘right’ methyl group
FY_d,FY_e,FY_z : Phenylalanine and Tyrosine H–C pairs at either the delta, epsilon, or zeta positions.
FY_d1,FY_e1,FY_z1:Same as above, but only one pair returned for each amino acid
We can also filter based on residues, segments, and a filter string (MDAnalysis format).
if topo is not None and Nuc is not None:
data.select=pyDR.MolSelect(topo=topo)
data.select.select_bond(Nuc=Nuc,resids=data.label,segids=None)
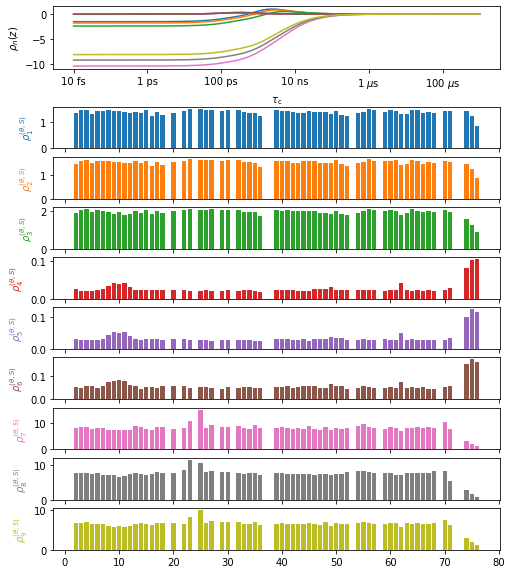
Plot the data#
plt_obj=data.plot(style='bar')
plt_obj.fig.set_size_inches([8,10])
Process NMR data#
data.detect.r_auto(n) #Set number of detectors here
fit=data.fit() #Fit the data
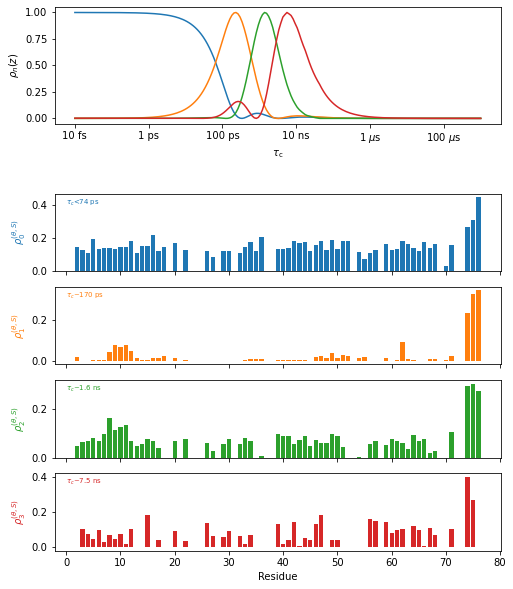
Plot the results#
proj.close_fig('all')
plt_obj=fit.plot(style='bar')
plt_obj.fig.set_size_inches([8,10])
plt_obj.show_tc()
_=plt_obj.ax[-1].set_xlabel('Residue')
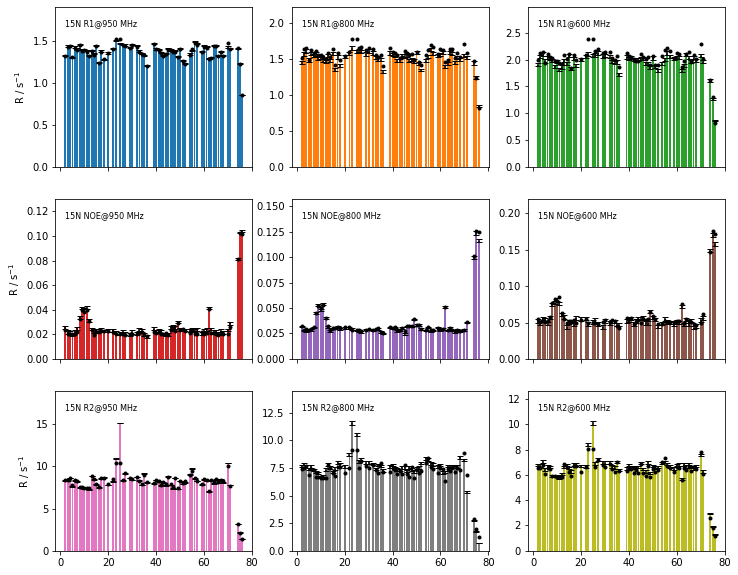
Plot the fit quality#
fig=fit.plot_fit()[0].axes.figure
fig.set_size_inches([12,10])
Visualize with NGL viewer#
fit.nglview(1,scaling=None)
Visualize with ChimeraX#
fit.chimera()